Kinderzahnheilkunde 11.01.2017
Erkrankungen mit Beteiligung der Mundschleimhaut bei Kindern - Teil 2






share

Der erste Teil (Oralchirurgie Journal 3/2016) gab einen kleinen Überblick über Erkrankungen mit Beteiligung der Mundschleimhaut bei Kindern. Gerade bei Kleinkindern spielen virale, bakterielle und mykotische Erkrankungen eine große Rolle, da alles – egal, ob Gegenstände auf der Straße oder Spielzeuge von anderen Kindern – mit dem Mund abgetastet wird. Neben den typischen infektionsbedingten Mundschleimhauterkrankungen gibt es auch nicht infektionsbedingte Erkrankungen sowie Systemerkrankungen, bei denen die Mundschleimhaut beteiligt ist. Teil 2 rundet die Ausführungen der letzten Ausgabe ab.
Herpangina (Zahorsky)
Die Herpangina wird vor allem durch das Coxsackie-Virus A Typ 4 hervorgerufen. Betroffen sind üblicherweise Kinder bis zum 4. Lebensjahr. Typischerweise kommt es nach einer Inkubationszeit von weniger als einer Woche zu plötzlich auftretendem Fieber, Übelkeit, Erbrechen, reduziertem Allgemeinzustand sowie Muskelschmerzen.36 Nach wenigen Stunden kommt es im Bereich vom Oropharynx zu heftigen Halsschmerzen und Schluckbeschwerden. Am Weichgaumen und den Gaumenmandeln entstehen steck- nadelkopfgroße Bläschen mit einem roten Halo (Abb. 1). Nach einem Tag platzen die Bläschen und lassen fibrinbelegte Erosionen zurück. Nach einer Woche klingen die Beschwerden wieder ab. Die Diagnose ergibt sich klinisch und lässt sich von der Gingivostomatitis herpetica vor allem durch die fehlende Gingivabeteiligung abgrenzen.37







Morbus Heck (fokale epitheliale Hyperplasie)
Bei der fokalen epithelialen Hyperplasie handelt es sich um eine Erkrankung der oralen Mukosa, die sich multilokulär an der Mundschleimhaut manifestiert. Sie wurde erstmals 1965 als eigenständige Entität beschrieben.38
Klinisch imponieren multiple, rund-ovale, einzeln oder konfluierende, leicht erhabene oder knotig geballte Papeln von weicher Konsistenz. Bei breitblasigem Aufsitzen erreichen sie in der Regel eine Größe von maximal 1 cm. Die Oberfläche ist meist fein gestippt und erscheint durch eine leichte Verhornung schwach weißlich. Die Schleimhautproliferationen befinden sich an der Lippenschleimhaut, an der Wangenschleimhaut, an Zungenrücken und Zungenrand sowie an der Gingiva des Frontzahnbereichs. Die Abbildungen 2a und b zeigen jeweils viral bedingte Akanthome, denen sich DNA von HPV 13 nachweisen ließ.
Befallen sind vorwiegend Kinder, es werden aber auch Erwachsene beiderlei Geschlechts betroffen. Bei Eskimos in Grönland, Kanada und Alaska sowie bei Indianern in Nord- und Südamerika ist die Erkrankung endemisch. Auch in Südafrika und der Türkei tritt die Krankheit auf. In der Bundesrepublik Deutschland ist sie sehr selten. Im Zuge der derzeit starken Immigration aus südlichen Erdteilen kann auch in Zukunft bei uns mit einem häufigeren Auftreten gerechnet werden.39
Ätiologisch gilt heute die virale Genese durch den mehrfach gelungenen Nachweis krankheitsspezifischer humaner Papillomviren als gesichert (besonders die Typen 13 und 32). Genetische Faktoren sind nicht bekannt.40
Die schmerzlosen Schleimhautveränderungen entsprechen dem Typ eines Papilloms und können sich nach jahrelangem Bestehen spontan zurückbilden. Differenzialdiagnostisch ist unter anderem an Verrucae vulgares und Condylomata accuminata zu denken. Die Erkrankung tritt auch als opportunistische Begleitinfektion bei erworbener Immunschwäche auf.39
Eine Biopsie zur Absicherung der klinischen Diagnose sollte stets unternommen werden, insbesondere, um in Anbetracht der möglichen Spontanremission unnötige radikale chirurgische Maßnahmen zu vermeiden. Zur Behandlung kommen therapeutisch isolierte Kürettagen oder Laserexzisionen in Betracht. Alternativ kann auch eine Therapie mit Imiquimod, einem Immunmodulator, stattfinden.41,42
Intraorale Hämangiome und vaskuläre Malformationen
Der Begriff Hämangiom wird traditionell genutzt, um eine Vielzahl von vaskulären Anomalien zu beschreiben. Im Gegensatz zu vaskulären Malformationen, bei denen es sich um strukturelle Anomalien der Blutgefäße ohne endotheliale Proliferation handelt, werden Hämangiome als benigne Tumoren angesehen, die eine schnelle Endothelproliferation zeigen. Während Hämangiome bei Geburt vorliegen oder sich innerhalb der ersten acht Lebenswochen entwickeln, bestehen vaskuläre Malformationen definitionsgemäß bereits bei Geburt und persistieren das gesamte Leben über. Je nach involvierten Gefäßen teilt man sie in kapilläre, venöse oder arteriovenöse Malformationen mit hohem oder niedrigem Blutfluss ein (Abb. 3; Hämangiom Oberlippe). Hämangiome stellen die häufigsten Tumoren im Kindesalter dar, sie treten bei 5 bis 10 % aller einjährigen Kinder auf, wobei Mädchen drei- bis fünfmal häufiger betroffen sind.43
Selten bestehen Hämangiome in vollständiger Ausprägung bereits bei der Geburt. Typischerweise entwickeln sie sich über einen Zeitraum von sechs bis zehn Monaten. Danach verlangsamt sich das Wachstum oder es kommt zur Persistenz ohne Wachstum. Bis zum fünften Lebensjahr hat sich rund die Hälfte aller Hämangiome spontan zurückgebildet, im Alter von neun Jahren sind 90 % der Hämangiome wieder verschwunden. Die Hämangiome stellen sich rötlich bis blaurot, erhaben oder leicht vorgewölbt dar. Bei Druck verblassen sie.
Die häufigste Komplikation von Hämangiomen stellt mit 20 % die Ulzeration mit sekundärer bakterieller Infektion dar. Anders als erwartet sind signifikante Blutungen äußerst selten. Bei Hämangiomen in speziellen Lokalisationen, wie z. B. der Orbita, kann es zu schweren Komplikationen, wie einer Amblyopie durch Druckschädigung kommen. Große zervikofaziale Hämangiome kommen auch im Rahmen des PHACE(S)-Syndroms vor:44
- Posteriore Gehirnanomalie (meistens Dandy-Walker-Syndrom)
- Hämangiome (zervikofazial lokalisiert)
- Arterielle Anomalien
- Cardiale Defekte (meist Coarctatio aortae)
- Eye (Augen) Anomalien
- Sternale Spalte
Ein weiteres, seltenes Syndrom, das mit dem Auftreten von Hämangiomen assoziiert wird, ist das Kasabach-Merritt-Syndrom. Hierbei handelt es sich um eine ernste Koagulopathie. Häufig kommt es zur Thrombozytopenie und Blutungen durch Plättchenverbrauch im Hämangiom. In 20 bis 30 % der Fälle kommt es zu einem tödlichen Ausgang.45
Vaskuläre Malformationen hingegen bestehen bereits bei Geburt und bilden sich auch nicht zurück. Die Prävalenz liegt bei 0,3 bis 1 % aller Neugeborenen. Venöse Malformationen schimmern typischerweise bläulich und schwellen bei zunehmendem venösem Druck an. Sekundär kommt es häufig zu Thrombosen oder zur Ausbildung von Phlebolithen. Arteriovenöse Malformationen sind durch einen hohen Blutfluss mit einer direkten Verbindung von Arterien und Venen gekennzeichnet.43
Da sich die meisten Hämangiome spontan zurückbilden, ist hier meistens ein abwartendes Vorgehen angezeigt. Außer bei lebensbedrohlich großen Hämangiomen oder bei starker Wachstumstendenz ist im Kindesalter selten die Indikation zur chirurgischen Therapie gegeben. Bei systemischer Kortisongabe kommt es in 70 bis 90 % zu einem kompletten Ansprechen der Hämangiome.46 Für vaskuläre Malformationen stellen gepulste Farbstofflaser eine Behandlungsalternative dar.47
Lues connata
Syphilis ist eine weltweit vorkommende Geschlechtskrankheit mit zunehmender Tendenz auch wieder in Deutschland. Der natürliche Wirt ist der Mensch. Erreger dieser meldepflichtigen Infektionserkrankung ist Treponema pallidum ssp. pallidum (Abb. 4). Bei Erwachsenen dringen die Erreger über Haut und Schleimhäute in den Körper ein, jedoch ist auch eine diaplazentare Übertragung möglich, die zur Manifestation der Erkrankung bei Säuglingen führt.48
Eine diaplazentare Übertragung ist ab dem vierten Schwangerschaftsmonat möglich. Bei hohen Erregerzahlen kann es vom sechsten bis achten Monat zum Spätabort kommen. Es wird bei Kindern zwischen der Lues connata praecox, die bereits bei Neugeborenen auftritt und in den ersten beiden Lebensjahren zu Symptomen führt, und der Lues connata tarda, bei der es erst in späteren Jahren zu sichtbaren Zeichen (sogenannte Stigmata) kommt, unterschieden.
Symptome der Lues connata praecox sind eine Hepatosplenomegalie, ein Ikterus, ein kutanes Pemphigoid und der syphilitische Schnupfen (Coryza syphilitica). Die Lues connata tarda ist durch die Hutchinson-Trias gekennzeichnet, die aus den Tonnenzähnen, der Keratitis parenchymatosa und Innenohrschwerhörigkeit besteht.49 Bei den Tonnenzähnen handelt es sich um eine Fehlbildung der Zahnkronen, die vor allem die mittleren oberen Schneidezähne betrifft. Sie haben eine Tonnenform der Krone mit einer halbmondförmigen Einbuchtung der Inzisalkante. Weiterhin können die unteren mittleren Schneidezähne und auch die ersten Molaren betroffen sein. Weitere Stigmata der Lues connata tarda sind die Säbelscheidentibia, eine Sattelnase, einer hoher gotischer Gaumen und die Parrot-Furche (perioral zirkulär angeordnete Rhagaden, die bis ins Lippenrot reichen).49
Morbus Kostmann (Kostmann-Syndrom, Infantile Agranulozytose)
Beim Kostmann-Syndrom handelt es sich um eine seltene autosomal-rezessive Störung der Granulopoese mit Fehlen aller Reifungsstadien, die zu einer massiven Anfälligkeit gegenüber bakteriellen Infektionen führt. Vor Einführung der Knochenmarktransplantation und insbesondere der G-CSF-Therapie (Granulozyten-Kolonie-stimulierender Faktor) führte die Erkrankung im frühen Säuglingsalter zum Tod.50,51
Aufgrund der eingeschränkten Immunabwehr kommt es häufig ab dem zweiten Lebensjahr als intraorale Manifestation zu erosiven Gingivitiden und einer rasch progredienten Parodontitis marginalis profunda. Dieses Beschwerdebild verläuft in der Regel in schubweisen Exazerbationen und führt zu bleibenden Schäden am jugendlichen Gebiss mit fortschreitendem Knochenabbau, Zahnlockerungen und Zahnfehlstellungen.52
Leukämien
Leukämien sind bösartige Erkrankungen, die durch autonome, irreversible Wucherungen leukozytärer Zellen (Abb. 5) und Verdrängung der normalen Hämatopoese im Knochenmark definiert sind. Die Krankheit ist als maligner Tumor des blutbildenden Systems anzusehen, früh werden Milz, Leber, Lymphknoten und auch andere Organe von den Leukämiezellen befallen.
Es handelt sich um eine heterogene Krankheitsgruppe, deren Ätiologie noch weitgehend ungeklärt ist. Nach der klinischen Symptomatologie, dem Krankheitsverlauf, dem Reifegrad und der Abstammung der Zellen werden akute (unreifzellige) und chronische (reifzellige) Leukämien unterschieden und hierbei in myeloische und lymphatische Formen eingeteilt.53
Im Rahmen der zahnärztlichen Diagnostik interessieren besonders die akuten Leukämien, da diese klinisch oft sehr früh Veränderungen an der Mundschleimhaut und besonders an der Gingiva als sichtbare Wegweiser für die Krankheit aufzeigen.54
Da eine detaillierte Besprechung der verschiedenen Leukämieformen den Rahmen dieses Artikels sprengen würde, werden hier nur die häufigsten Befunde dargestellt. Der Interessierte sei auf die einschlägige internistisch-hämatologische Literatur verwiesen.
Akute Leukämien (akute Leukosen, unreifzellige Leukämien)
Unbehandelt führen diese innerhalb von Wochen bis wenigen Monaten zum Tode. Sie sind durch das Auftreten von unreifen Zellen des hämatopoetischen Systems („Blasten“ im Knochenmark) und peripheren Blut gekennzeichnet. Die Erkrankung kann auf andere Körpergewebe übergreifen. Die Inzidenz beträgt in der Bundesrepublik Deutschland 6/100.000 Menschen jeden Lebensalters.
Es wird zwischen der akuten myeloischen Leukämie (akute Myeloblastenleukämie, AML), die vornehmlich im Erwachsenenalter auftritt, und der akuten lymphatischen Leukämie, die mit zwei Häufigkeitsgipfeln zunächst im Kindesalter (zweites bis sechstes Lebensjahr) und erneut mit rasch zunehmender Häufigkeit im höheren Lebensalter auftritt, unterschieden.
Die Allgemeinsymptomatik akuter Leukämien tritt meist plötzlich aus voller Gesundheit heraus auf, mit Zeichen eines schweren fieberhaften Infekts und zunehmender Blutungsneigung aus Mund, Nase, Magen-Darm-Trakt und den ableitenden Harnwegen, aufgrund einer thrombozytopenischen Blutungsneigung. Die intraorale Symptomatik der akuten Leukämie zeigt nahezu regelmäßig und oft schon im Frühstadium Veränderungen in der Mundschleimhaut, vor allem an der Gingiva:54,55
Als Leitsymptome gelten:
- Nekrosen-Ulzerationen
- Gewebsvermehrung der Gingiva (durch leukämische proliferative Infiltrate), keine Gingivahyperplasie
- Blutungen (meist thrombozytopenischer Natur, seltener plasmatische hämorrhagische Diathesen)
Häufig kommt es infolge der Abwehrschwäche zu Candidiasis der Mundhöhle, die als opportunistische Infektion zu deuten ist. Bei der akuten lymphatischen Leukämie im Kindesalter sind ausgeprägte Gingivaveränderungen und Veränderungen des Parodontiums selten. Typischerweise liegen bei den akuten lymphatischen Leukämien regelmäßig Vergrößerungen der Halslymphknoten vor. Infolge der hämorrhagischen Diathese ist eine Blutungsneigung unterschiedlichsten Ausmaßes möglich: von leichten Ekchymosen bis zu profusen Schleimhautblutungen. Der diagnostische Stellenwert dieser in wechselnder Kombination auftretenden intraoralen Kardinalsymptome ist immer in Zusammenhang mit den Allgemeinerscheinungen zu sehen.54,55
Hierbei ist stets zu berücksichtigen, dass die Veränderungen in der Mundhöhle in Abhängigkeit von Art und Verlaufsform der Leukämie, dem Ausmaß der Resistenzschwäche und dem Alter der Patienten außerordentlich variabel sind. Es muss betont werden, dass kein Gingiva-, Parodontal- und Mundschleimhautbefund pathognomonisch für eine bestimmte Leukämieform ist. Allgemein gilt festzustellen, dass akute Leukosen im Erwachsenenalter häufiger zu intraoralen Manifestationen führen und mit schwereren Veränderungen in der Mundhöhle einhergehen als die chronischen Leukämien. Für den Zahnarzt ergibt sich hieraus eine besondere diagnostische Herausforderung, die in der frühzeitigen Äußerung der Verdachtsdiagnose einer akuten Leukämie liegt, die wiederum per se zu einer raschen Veranlassung weiterer Abklärung führen wird.56
Fazit: Eine akut ulzerierende oder akut nekrotisierende Gingivostomatitis ist, bei Behandlungsresistenz über einige Tage hinweg, als dringend verdächtig auf eine maligne Systemerkrankung anzusehen und sollte, um keine wertvolle Zeit für die Behandlung eines solchen Krankheitsbildes zu verlieren, zeitnah hämatologisch abgeklärt werden.
Hyperkeratosis palmoplantaris (Papillon-Lefèvre-Syndrom)
Das Papillon-Lefèvre-Syndrom ist eine seltene autosomal-rezessiv vererbte Erkrankung, die zur Gruppe der seltenen Palmoplantarkeratosen gehört. Sie führt zu einer übermäßigen Verhornung (Hyperkeratose) an Händen und Füßen und geht mit schweren Schädigungen der parodontalen Gewebestrukturen einher. Die Hyperkeratosen treten gewöhnlich zwischen dem ersten und dritten Lebensjahr auf.57
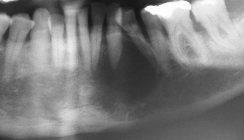
Die intraorale Manifestation zeigt parallel zu den Verhornungsstörungen schwere entzündliche Veränderungen am Zahnstützgewebe, schon kurz nach dem Durchbruch der Milchzähne. Es kommt zu akuten Entzündungen der Interdentalpapillen und der marginalen Gingiva, die zur Bildung von Zahnfleischtaschen führen, aus denen sich Pus entleert. In kurzer Zeit entwickelt sich eine Parodontitis marginalis profunda, die je nach Ausprägungsgrad des Syndroms zu einem Ausfallen der Milchzähne bis zum vierten oder fünften Lebensjahr führt.58 Im zahnfreien Intervall zwischen dem Verlust der Milchzähne und dem Durchbruch der bleibenden Zähne ist die Alveolarmukosa weitgehend unauffällig. Nach Durchbruch der bleibenden Zähne wiederholt sich der oben beschriebene parodontale Destruktionsprozess, sodass auch die bleibenden Zähne bis zum 15. Lebensjahr verloren gehen. Die Weisheitszähne bleiben verschont.52,58 Röntgenaufnahmen zeigen analog zum klinischen Befund massive Osteolysen der alveolären Knochenstrukturen.59
Die zahnärztliche Therapie kann bestenfalls den Ablauf des parodontalen Destruktionsprozesses protrahieren. Neuere klinische Erfahrungen sprechen für den positiven Einfluss von Retinoiden, nicht nur auf die Hyperkeratosen, sondern auch auf die marginale Parodontitis.60,61
Kawasaki-Syndrom (mukokutanes Lymphknotensyndrom)
Das Kawasaki-Syndrom ist eine akute, fieberhafte (lang anhaltendes Fieber) systemische Erkrankung, die durch eine nekrotisierende Vaskulitis der kleinen und mittleren Arterien gekennzeichnet ist.62 Es kommt zu einer Entzündung in vielen Organen, die mit Ikterus, Arthralgien, Meningitis, Schwellungen der Halslymphknoten, polymorphen Exanthemen, Plantar- und Palmarerythemen, Konjunktivitis und Mundschleimhautbeteiligung einhergeht; die Zunge kann wie bei Scharlach im Sinne einer Himbeerzunge verändert sein.63 Das Kawasaki-Syndrom betrifft vor allem Kleinkinder und imitiert im anfänglichen Erscheinungsbild Infektionskrankheiten wie Masern oder Scharlach.64
Die orale bzw. intraorale Manifestation umfasst das Bild einer trockenen Cheilitis, die oben genannte Himbeerzunge, sowie eine intensive Gingivostomatitis mit Rötung und Schwellung. Diese Veränderungen dauern in der Regel so lange wie das Fieber persistiert.63 Eine für das frühe Lebensalter ungewöhnliche Komplikation ist die Bildung von Aneurysmata, Stenosen oder Thromben in den Koronargefäßen (Abb. 6) in einzelnen Fällen.62,65
Die vollständige Literaturliste gibt es hier.